IDMP Definition of Identification of Medicinal Products
IDMP defines the data elements and structures for the unique identification and exchange of medicinal products´ information.
In June 2017, the first two so-called “Implementation Guides” for the implementation of the ISO IDMP data standards have been published and put into force. It is the “Implementation Guide” for organizational characteristics of marketing authorization holders (OMS – Organization Management Service) and the “Implementation Guide” for EMA’s lists of terms (RMS – Referential Management Services), also known as the so-called “Controlled Vocabularies”.
IDMP Standards – What is IDMP?
New regulations for the identification of medicinal products are currently being actively processed by the EMA, the FDA and most likely by other authorities as well: According to Article 57 (2) of Regulation (EC) No. 726/2004 and Article 40 of the Implementing Regulation (EC) 520/2012, the European Commission has determined the introduction of ISO IDMP (Identification of Medicinal Products) data standards as of 01.07.2016 as the regulatory electronic exchange format of product information for all medicinal products approved (CP, MRP, DCP or NP) in Europe.
A common database is intended to facilitate the identification of drugs and active ingredients. This is of importance for pharmacovigilance and regulatory activities related to medicinal products, e.g. to bundle adverse reaction reports across Europe and thus to be able to reduce response times with regard to drug monitoring.
Pharmaceutical companies in Europe must mandatorily implement these legal regulations and are affected by a gradual introduction, which means a large to very large effort (depending on the number of existing medicinal products).
Since the end of 2015, the EMA has been working on the transition from the previous so-called “Preparation Phase” to the “Transition Phase” for IDMP. For the start of this phase, the above-mentioned “Implementation Guides OMS and RMS” were published by June 2017.
The two further Implementation Guides, the Substance Management Service and the Product Management Service, will be published in mid-2018, according to the current planning of the EMA as per February 2017. In the further course of the “Transition Phase”, companies are expected to submit extensive electronic data on their products to the EMA in four steps over several years (2018 – 2024).
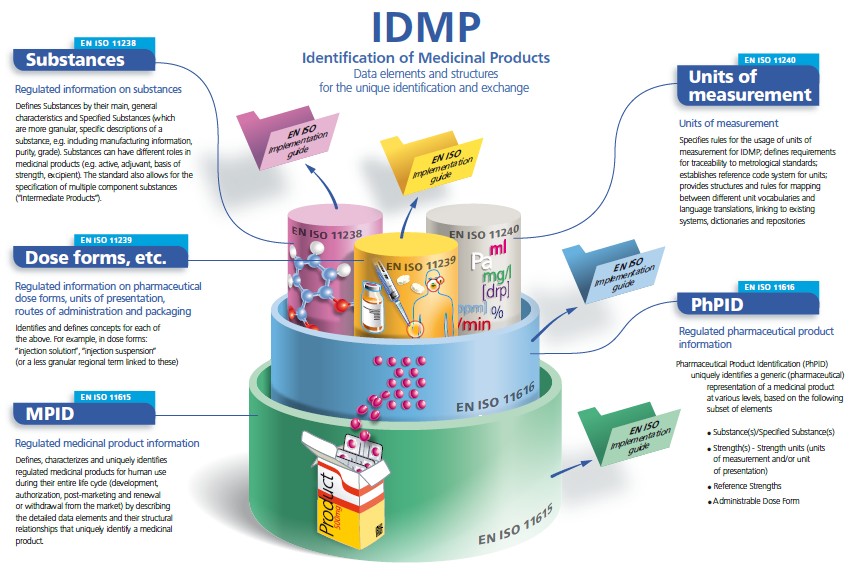
ISO IDMP Standards
The set of fife ISO international standards has been developed by the ISO in response to a worldwide demand for internationally harmonised specifications for identification and description of medicinal products.
Article 40 of the new pharmacovigilance implementing measures legislation mandated the EMA to use the ISO IDMP standard latest by July 2016.
IDMP provides the basis for the unique identification of medicinal products, which facilitates the activities of medicines regulatory agencies worldwide by jurisdiction for a variety of regulatory activities (development, registration and life cycle management of medicinal products, pharmacovigilance, and risk management). They can also be applied to Investigational Medicinal Products.
Messaging specifications are included as an integral part of the IDMP Standards. They describe and protect the integrity of the interactions for the submission of regulated medicinal product information in the context of the unique product identification; they include acknowledgement of receipt including the validation of transmitted information. Health Level Seven (HL7) Message Exchange are normative within the IDMP Standards.
IDMP Standards are completed with Implementation Guides which are currently in development (2015), as well as with Technical Specifications (TS) 16791 (provides guidance for the identification of medicinal products by using intenational supply chain Standards, securing traceability, safe supply chain and other market requirements) and Technical Requirements (TR) 14872 (Requirements for the implementation of the Standards for the identification of medicinal products for the exchange of regulated medicinal product Information), the latter being in development.
Implications for Marketing Authorisation Holders (MAHs)
In order to submit the data all branches of a company should share a common language, from pharmacovigilance to product supply to comply with the controlled vocabulary dedicated.
The structured substance information, and controlled vocabularies for pharmaceutical dose forms, units of presentation, routes of administration, and packaging will be challenging to integrate.
Furthermore, the terminology needs to be aligned throughout the company.